Alexandre de Brevern - Thčse de Bioinformatique Moléculaire
L'objectif de ce travail est d'obtenir un grand nombre de prototypes pour pouvoir ensuite reconstruire l'ensemble des structures protéiques avec ces blocs (1989,[198] et 1993,[200]). Pour leur base de données, Unger et collaborateurs ont conservé sur les 354 chaînes présentes (PDB de janvier 1987), 82 ayant un facteur R dit "correct" et une résolution de moins de 2.8 Å, soit 12 973 résidus.
Leur méthode consiste à calculer l'écart
quadratique moyen ou RMSd (root mean square deviation) entre deux structures s
et t, en ne conservant que les carbones ![]() du squelette
dans cette comparaison:
du squelette
dans cette comparaison:
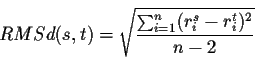
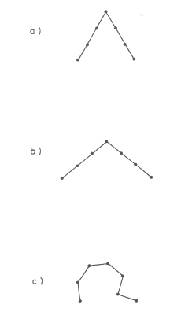
Un exemple intéressant des problèmes liés au calcul du RMSd est donné. Le RMSd minimal entre deux structures n'est pas obligatoirement une information adéquate. Ainsi la figure 2.3.2.1 montre 3 fragments protéiques. Le RMSd entre (a) et (b) est plus grand que celui entre (b) et (c), alors que le type de repliement de (a) est plus proche de celui de (b).
Après un calcul préliminaire effectué sur 4 protéines (426 résidus au total), Unger et collaborateurs ont décidé de prendre comme prototypes des hexamères (i.e., des fragments de longueur 6). Ils considérent cette longueur comme la plus petite correcte pour bien différencier les fragments protéiques entre eux. L'ensemble des RMSd entre les hexamères des 4 protéines en question (soit 82 215 couples) est calculé et ensuite réuni par une méthode dite "d'annexion".
Cette procédure se passe en trois étapes:
La méthode d'annexion est moins conventionnelle que la méthode des nuées dynamiques. La plus grande crainte des auteurs est de n'obtenir que fort peu de groupes dans la première étape. Elle donne 55 groupes, ce nombre étant directement lié au choix de leur valeur limite de 1 Å , qui est assez faible pour des fragments de cette taille. La seconde étape est plus criticable. En effet, rien ne laisse supposer qu'un groupe X doit être subdivisé en sous-groupe X1 et X2. Il est logique de penser que des éléments de X après division puissent être alors plus proches d'un autre groupe Y plutôt que X1 ou X2. Après la seconde étape, 103 blocs structuraux sont obtenus. Sur la base de données complète :
Utilisant 4 autres protéines, la méthode donne à 144 blocs protéiques finaux. En combinant les 8 protéines, elle atteind à 170 blocs structuraux. Les plus fréquents sont retrouvés dans les deux expériences, mais le reste est trés fluctuant. Les structures connues répétitives et leurs liens caractéristiques sont retrouvés.
Une méthode de reconstruction sur des protéines de longueur 60, ou alors sur les 60 premiers acides aminés (extrémité N-terminale), est effectuée en ajoutant à chaque fois "au mieux" un nouveau carbone correspondant au nouveau bloc protéique. La procédure consiste à superposer en une position X les 5 premiers atomes du bloc structural x avec les 5 derniers de la chaîne reconstruite. La reconstruction est correcte dans plus de 64% des cas. Seuls des changements rapides dans le repliement posent un réel problème.
En conclusion, la méthode développée par Unger et collaborateurs est intéressante car elle est basée sur une méthodologie pertinente pour regrouper des fragments protéiques, et surtout, les blocs ont servi lors d'une méthode de reconstruction 3D avec de bons résultats. Toutefois, deux points posent problémes :
L'équipe d'Unger utilisera ces blocs, comme exemple, pour une autre méthode de reconstruction, basée sur les angles dièdres [199]. Ils montrent que la plupart des hexamères trouvés sont créables par une procédure aléatoire dans les zones de Ramachandran classiques.
Enfin en 1993, Unger et Sussman réutilisent les blocs obtenus quatre ans auparavant [200],
mais n'en conservent que 81,
ceux-ci étant observés plus de 35 fois dans leur base de données, soit une fréquence minimale de 0,3 %.
Détail troublant, les chiffres donnés
sont les même que ceux obtenus pour les 103 blocs.
Les hexamères obtenus se retrouvent dans les zones préférentielles du
diagramme de Ramachadran, mais beaucoup sont significatifs car une procédure purement aléatoire
crée beaucoup de fragments peu ou non observés dans la base de données.
Ainsi, les auteurs espèrent déceler un début d'architecture des protéines avec leurs hexamères.
Pour explorer un cas plus localisé,
tous les blocs définis
en 'brin étendu' ('E' ou feuillet ![]() dans DSSP [100])
sont extraits de la base de données.
Ces différents blocs ont une répartition différente en acides aminés. Toutefois, les
chiffres donnés sont en pourcentage et on ne peut juger de la valeur statistique
des variations observées.
dans DSSP [100])
sont extraits de la base de données.
Ces différents blocs ont une répartition différente en acides aminés. Toutefois, les
chiffres donnés sont en pourcentage et on ne peut juger de la valeur statistique
des variations observées.
(c) 2001- Alexandre de Brevern