Alexandre de Brevern - ThŤse de Bioinformatique Molťculaire
L'objectif des travaux de Schuchhardt et collaborateurs est d'obtenir un grand nombre de prototypes pour approximer la structure tridimensionnelle d'une protéine dans un but de classification et d'analyse sans tenter de reconstruire cette structure (1996, [175]).
La base de données est composée de 136 protéines ayant moins de 30% de similitude de séquence [84,83], ce qui représente un total de 24 239 résidus.
La méthode consiste à caractériser les protéines en utilisant des fragments d'une longueur de
9 résidus décrits par les angles ![]() et
et ![]() . Les protéines sont découpées
en fragments de 9 résidus donnant chacun un vecteur d'observation de longueur 16
(le premier
. Les protéines sont découpées
en fragments de 9 résidus donnant chacun un vecteur d'observation de longueur 16
(le premier ![]() et le dernier
et le dernier ![]() n'étant pas pris en compte).
L'ensemble comporte donc 23 151 exemples.
Schuchhardt et collaborateurs ont utilisé
une méthode non supervisée d'apprentissage, les cartes de Kohonen (cf. Annexe 1 pour plus de dťtail).
Cette méthode consiste à créer N
n'étant pas pris en compte).
L'ensemble comporte donc 23 151 exemples.
Schuchhardt et collaborateurs ont utilisé
une méthode non supervisée d'apprentissage, les cartes de Kohonen (cf. Annexe 1 pour plus de dťtail).
Cette méthode consiste à créer N ![]() M neurones (représentés par une matrice),
chaque neurone étant une observation moyenne, ici un vecteur d'angles diŤdres,
à rechercher pour chaque observation de la base de données le neurone le plus
proche de cette observation et lui faire apprendre cette observation.
Le principe est décrit en détail dans l'annexe I. Son intérêt principal est un
aspect visuel particulièrement utile pour l'analyse.
La méthode des cartes de Kohonen ne diffère des nuées dynamiques ou des k-moyennes (ou k-means) [79]
que par l'existence d'un processus de diffusion qui permet de réunir dans un environnement
proche des neurones ayant des points communs.
M neurones (représentés par une matrice),
chaque neurone étant une observation moyenne, ici un vecteur d'angles diŤdres,
à rechercher pour chaque observation de la base de données le neurone le plus
proche de cette observation et lui faire apprendre cette observation.
Le principe est décrit en détail dans l'annexe I. Son intérêt principal est un
aspect visuel particulièrement utile pour l'analyse.
La méthode des cartes de Kohonen ne diffère des nuées dynamiques ou des k-moyennes (ou k-means) [79]
que par l'existence d'un processus de diffusion qui permet de réunir dans un environnement
proche des neurones ayant des points communs.
Pour évaluer les différences entre les observations, une mesure de dissemblance, le RMSda ou root mean square deviation on angular values est utilisée. Cette distance sur les angles a déjà servi dans une méthode de recherche d'homologie structurale [102]. Il s'agit d'une distance euclidienne entre deux vecteurs d'observations s et t:
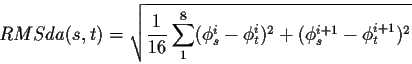
La figure 2.12 montre la carte de Kohonen obtenue pour une taille
de 10 ![]() 10, soit 100 neurones. Cette carte est "plane", ce qui veut dire
qu'elle possède des bords, le neurone (9/9) n'est voisin
que des neurones (8/8), (8/9) et (9/8), alors qu'au milieu de la carte, un neurone est entourť de 8
voisins.
10, soit 100 neurones. Cette carte est "plane", ce qui veut dire
qu'elle possède des bords, le neurone (9/9) n'est voisin
que des neurones (8/8), (8/9) et (9/8), alors qu'au milieu de la carte, un neurone est entourť de 8
voisins.
 |
Les feuillets et les hélices se retrouvent dans des neurones distincts.
Les hélices ![]() sont fortements localisées en haut et à gauche de la carte,
les feuillets
sont fortements localisées en haut et à gauche de la carte,
les feuillets ![]() plus sur la droite.
La validation structurale est effectuťe exclusivement sur le critère du RMSda
et les rťsultats semblent satisfaisants.
Des motifs dits de "cassure" classique avec une forte proportion de Glycine sont trouvés.
Quelques exemples de suivi de trajectoires de protéines sur la carte sont montrés.
plus sur la droite.
La validation structurale est effectuťe exclusivement sur le critère du RMSda
et les rťsultats semblent satisfaisants.
Des motifs dits de "cassure" classique avec une forte proportion de Glycine sont trouvés.
Quelques exemples de suivi de trajectoires de protéines sur la carte sont montrés.
La méthode est particulièrement appropriée à la recherche et à l'analyse de données biologiques, comme le montre le travail de Hanke et Reich sur la reconnaissance de motifs propres à des familles protéiques [77,78]. L'utilisation des angles diŤdres avec le RMSda est beaucoup plus souple que le classique RMSd. Du fait de sa rapiditť, un travail plus important sur les diffťrents paramètres de l'apprentissage est possible. Les résultats sont convaincants. Toutefois, ayant refait leurs expériences, deux points me paraissent importants :
(c) 2001- Alexandre de Brevern