Figure 2.13:
Représentation des angles des 4 blocs de (a) longueur 4, (b) longueur 5, (c) longueur 6 et
(d) longueur 7 (recalculés à partir de la figure3. p.331-332 [162]).
Sur les 4 classes observées pour chaque longueur, la famille 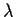 représente
toujours un ensemble exclusivement boucles, la famille
représente
toujours un ensemble exclusivement boucles, la famille 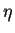 étant
composée d'hélices
étant
composée d'hélices 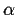 , la famille
, la famille 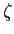 de boucles et de feuillets
de boucles et de feuillets  et
la famille
et
la famille 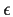 de feuillets .
Cette classification hiérarchique
est utilisée ensuite pour discriminer différentes familles protéiques.
La figure 2.13 donne les valeurs des angles
de feuillets .
Cette classification hiérarchique
est utilisée ensuite pour discriminer différentes familles protéiques.
La figure 2.13 donne les valeurs des angles
 et
et 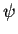 obtenues.
Je les ai caculées à partir de la figure 3 p.333 [162] pour pouvoir comparer mon alphabet avec le leur.
obtenues.
Je les ai caculées à partir de la figure 3 p.333 [162] pour pouvoir comparer mon alphabet avec le leur.
Dans un second temps [163],
le lien existant entre ces structures et les séquences a été recherché.
Pour cela, un travail important de formalisme de
la significativité d'une séquence a été effectué.
Chaque succession de 7 résidus a été traitée sous la forme
x - my - nz,
avec x, y et z des acides aminés déterminés compris dans une séquence
de longueur 7, avec n+m=4 (7-3), n et m variant donc, entre 1 et 3,
et représentant n'importe quel acide aminé.
Les occurrences conservées sont représentatives
à la fois de la séquence (un nombre d'occurrence supérieur à trois)
et la structure (associée majoritairement à un type de structure donnée).
Ce type de représentation avec des acides aminés non déterminés
vient de la taille limitée de la base de données [164] et d'un précédent travail
sur certaines formes de coudes [158].
Avec cette approche, les auteurs trouvent à partir
de la séquence, un taux de prédiction compris entre 41% et 47%.
Ce taux est à mettre en comparaison avec les taux des structures
secondaires de l'époque qui avoisinent 60% pour 3 états [62,65],
alors que les blocs en proposent 4.
En parallčle de cette observation, ils trouvent un plus grand
déterminisme dans les séquences observées avec leurs
4 blocs pour une longueur L=6,
mais en contre-partie un bruit de fond, lui aussi accentué.
Aucune amélioration de la prédiction n'est donc observée.
Ainsi, les blocs obtenus sont fortement liés
aux structures secondaires répétitives.
Le choix du nombre final de blocs peut porter à critiques.
Il ne permet pas, comme pour les structures secondaires,
de reconstruire directement une structure protéique à partir de cette information.
La taille de la base de données est pour beaucoup dans le
taux final de prédiction.
Par la suite, seule la méthode de prédiction, mais appliquée aux structures
secondaires, sera réutilisée [159] et mise en oeuvre dans
une recherche basée plus spécifiquement pour une recherche à partir de
coordonnées extraites de 7 régions dans la
carte de Ramachandran [159,160]
et avec utilisation de protéines homologues [161].
Prochain: Utilisation d'un réseau de
Au-dessus: Peu de blocs protéiques
Précédent: Peu de blocs protéiques
Page 32
(c) 2001- Alexandre de Brevern