Alexandre de Brevern - Thčse de Bioinformatique Moléculaire
Pour compacter notre base de données structurales, nous avons donc utilisé la méthode de la protéine hybride (cf. paragraphe 6.2). La protéine hybride est une protéine chimérique composée de N sites où chaque position i n'est pas définie par un seul bloc, mais par une loi de probabilité fi (bx), bx étant un des 16 BPs (x=1, 2,..., 16).
La figure 6.1 récapitule les différentes étapes de l'apprentissage. Dans un premier temps, la base de données de 553 protéines ayant moins de 25% de similitude de séquence (cf. paragraphe 3.3.2.6) établie par Romain Gautier a été traduite en terme de blocs protéiques.
Ensuite, l'apprentissage proprement dit consiste en une recherche de la position optimale de chaque fragment dans la protéine hybride (cf. figure 6.1c à 6.1f). L'apprentissage est proche des cartes auto-organisées (cf. Annexe 1), mais sans processus de diffusion. Ainsi, la protéine hybride correspond à des séries de structures locales "floues".
L'intérêt principal de la méthode est de maintenir la séquentialité pour créer des séries de structures locales successives ayant un fort taux de recouvrement. Cette stratégie diffère donc fortement d'une classification classique où les groupes sont indépendants.
Un fragment F est tiré aléatoirement dans la base de données (cf. figure 6.1c).
Chaque structure locale F est définie par L blocs consécutifs
![]() b1*,b2*,...,bL*
b1*,b2*,...,bL*![]() (ici L=10 BPs, soit 14 C
(ici L=10 BPs, soit 14 C![]() ).
).
Localisation de la structure locale dans la protéine hybride. Un score Si est calculé en chaque position i de la protéine hybride :
![\begin{displaymath}S_i=\sum_{k=1}^{k=L} ln[f_{i+k-1}(b_k^*)]\end{displaymath}](Pictures/img201.gif)
avec k=1,2,...,L.
Ainsi le score Si (i.e. le logarithme de la vraisemblance
d'observation de la structure locale F en un site donné i)
mesure la similitude entre la structure locale
et une région donnée de même dimension dans la protéine hybride (cf. figure 6.1d).
La région la plus proche de la structure locale possède le moins de différence avec celle-ci
et donc son score est maximal. Ainsi cette position i0 est définie comme (cf. figure 6.1e):
Modification locale de la protéine hybride. Ayant trouvé une zone de plus forte ressemblance, cette zone va être légèrement modifiée pour ressembler à la structure locale F. Les positions i0 à i0+L-1 seront ainsi modifiées (cf. 6.1f):
si b = bk* (i.e. le bloc protéique présent à la position k dans la structure locale), alors

si
![]() (i.e. les autres blocs protéiques présents à la position k), alors
(i.e. les autres blocs protéiques présents à la position k), alors
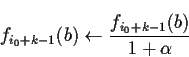
Le symbole
![]() signifie que la valeur calculée remplace la valeur précédente.
Le coefficient d'apprentissage
signifie que la valeur calculée remplace la valeur précédente.
Le coefficient d'apprentissage ![]() est encore égal à
est encore égal à
![]() ,
avec
,
avec
![]() le taux initial d'apprentissage (ici
le taux initial d'apprentissage (ici
![]() = 0.1),
t le nombre de structures locales de L blocs déjà utilisées dans
l'apprentissage et
= 0.1),
t le nombre de structures locales de L blocs déjà utilisées dans
l'apprentissage et ![]() le nombre
de structures locales présentes dans la base de données.
L'apprentissage est progressif, ainsi l'ensemble de la base de données a été examiné entièrement C fois,
ici C = 15
(phases: figure 6.1c à la figure 6.1f pour chaque structure locale de la base de données).
Un passage complet de la base de données est appelé cycle.
Pendant le premier cycle, le coefficient d'apprentissage
le nombre
de structures locales présentes dans la base de données.
L'apprentissage est progressif, ainsi l'ensemble de la base de données a été examiné entièrement C fois,
ici C = 15
(phases: figure 6.1c à la figure 6.1f pour chaque structure locale de la base de données).
Un passage complet de la base de données est appelé cycle.
Pendant le premier cycle, le coefficient d'apprentissage
![]() est maintenu constant (
est maintenu constant (![]() =
=
![]() ) pour modifier de façon importante la protéine hybride et ainsi
faire moins jouer l'initialisation [109].
) pour modifier de façon importante la protéine hybride et ainsi
faire moins jouer l'initialisation [109].
Initialisation. La protéine hybride est initialement définie par une série de N distributions sur les blocs fi(bx) quasiment identiques :
avec f(bx) la fréquence du bloc protéique bx dans la base de données,
![]() est une valeur aléatoire tirée dans l'intervalle [-
est une valeur aléatoire tirée dans l'intervalle [-![]() ;+
;+![]() ]
(
]
(![]() a été fixé à 0,20). En chaque position la somme des probabilités des blocs a été réajustée à 1,
en recalculant :
a été fixé à 0,20). En chaque position la somme des probabilités des blocs a été réajustée à 1,
en recalculant :
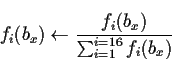
Il faut noter que la protéine hybride est "fermée" dans le sens où le Nème site est contigu avec le premier, ainsi, il n'existe pas d'effet de bord.
(c) 2001- Alexandre de Brevern