Alexandre de Brevern - Thčse de Bioinformatique Moléculaire
Le travail de Bystroff et Baker concerne l'obtention d'un certain nombre de blocs structuraux protéiques (I-sites) types pour faire de la prédiction. Ils cherchent ŕ optimiser la relation séquence-structure (1998, [19]). Les 471 protéines sont issues de la base de données HSSP [172], possèdent moins de 25% d'identités de séquences, et sont groupées en familles d'homologues.
Ce travail est ŕ l'heure actuelle le plus abouti et celui qui a généré le plus grand nombre d'applications. Les I-sites débutent en réalité trois ans avant leur réalisation par un travail de Han et Baker. Ils mettent au point une méthode de regroupement de séquences ayant de fortes similitudes de séquences par une méthode proche des k-means [74]. Dans un second temps, ils mettent en relation le fait que les groupes qu'ils ont obtenus peuvent correspondre ŕ un ou plusieurs types de repliements protéiques [75]. Ils se limitent ŕ une simple description de répartitions en structures secondaires. Ils définissent alors directement à partir de leur groupes certaines structures plus ou moins précises [76].
La figure 2.21 récapitule l'ensemble du processus de création des I-sites. Les 471 protéines issues de HSSP sont alignées en une centaine de groupes par un alignement multiple. Certaines protéines sont exclues si leur résolution est trop mauvaise, s'il y a plusieurs ponts disulfures ou si la protéine est membranaire. Les longues boucles semblent avoir été exclues aussi.
Ensuite un regroupement de fragments est effectué à l'aide de la méthode des k-moyens (ou k-means, cf. Annexe 1) avec l'utilisation de profils sur les acides aminés. K groupes dont les longueurs sont variables sont obtenus.
Ces K groupes sont ensuite regroupés suivant leur similitude de repliement. Cette similitude est mesurée par deux critéres :
Pour créer un groupe, on sélectionne les "meilleures" séquences de chacun des K groupes.
Ces séquences sont définies comme les plus proches du consensus (centre) de chaque groupe.
Deux fragments protéiques sont considérés structurellement similaires si leur
dme est inférieure ŕ 1.4 Å
et leur dma est
inférieure ŕ 120o. Cette méthode permet de regrouper leurs K groupes-séquences en K' groupes-structures.
Une nouvelle optimisation est alors effectuée: elle consiste ŕ ne conserver que les fragments
les plus proches sur le plan de la séquence et ŕ recalculer le profil séquence moyen.
Les 400 séquences les plus proches servent de nouveau centre et le processus
recommence. Il suffit de 3 ŕ 5 cycles d'apprentissage pour stabiliser les groupes.
Une autre étape d'optimisation suit, mais elle est peu décrite, et sert de dernier filtrage
entre la séquence et la structure.
Les figures 7.7 ŕ 7.11 montrent les K' = 13 groupes finaux obtenus ŕ partir des K = 82 groupes obtenus sur les séquences. Les figures présentant les I-sites sont mises en Annexe 3. Les tailles varient au départ entre 3 et 17 résidus. Après le second regroupement, les longueurs moyennes observées ne sont pas données.
Les groupes ou I-sites sont fortement liés aux structures secondaires répétitives avec pour les
hélices ![]() 7 sites : le I-site 1, une hélice
7 sites : le I-site 1, une hélice ![]() amphipatique (cf. figure 7.7a),
le I-site 2, hélice
amphipatique (cf. figure 7.7a),
le I-site 2, hélice ![]() non polaire (cf. figure 7.7b),
le I-site 3, extrémité C-terminale d'hélice
non polaire (cf. figure 7.7b),
le I-site 3, extrémité C-terminale d'hélice ![]() Glycine dite de type 1 (cf. figure 7.7c),
le I-site 4, extrémité C-terminale d'hélice
Glycine dite de type 1 (cf. figure 7.7c),
le I-site 4, extrémité C-terminale d'hélice ![]() Glycine dite de type 2 (cf. figure 7.8a),
le I-site 5, extrémité C-terminale d'hélice
Glycine dite de type 2 (cf. figure 7.8a),
le I-site 5, extrémité C-terminale d'hélice ![]() Proline (cf. figure 7.8b),
le I-site 6, hélice
Proline (cf. figure 7.8b),
le I-site 6, hélice ![]() mélée (cf. figure 7.8c),
le I-site 7, extrémité N-terminale d'hélice
mélée (cf. figure 7.8c),
le I-site 7, extrémité N-terminale d'hélice ![]() Sérine (cf. figure 7.9a),
pour les feuillets
Sérine (cf. figure 7.9a),
pour les feuillets ![]() 2 sites :
le I-site 8, feuillet
2 sites :
le I-site 8, feuillet ![]() amphipatique (cf. figure 7.9b)
le I-site 9, feuillet
amphipatique (cf. figure 7.9b)
le I-site 9, feuillet ![]() hydrophobe (cf. figure 7.9c),
pour les boucles 4 sites :
le I-site 10, coude
hydrophobe (cf. figure 7.9c),
pour les boucles 4 sites :
le I-site 10, coude ![]() Aspartate (cf. figure 7.10a),
le I-site 11, épingle
Aspartate (cf. figure 7.10a),
le I-site 11, épingle ![]() Sérine (cf. figure 7.10b),
le I-site 12, épingle type I étendu (cf. figure 7.10c),
le I-site 13, coude type II divergeant (cf. figure 7.11).
Sérine (cf. figure 7.10b),
le I-site 12, épingle type I étendu (cf. figure 7.10c),
le I-site 13, coude type II divergeant (cf. figure 7.11).
Plusieurs points donnent matière à discussion sur cette premičre partie. Tout d'abord,
l'importance des I-sites en relation avec
les hélices ![]() avec 3 I-sites pour la partie centrale,
1 pour les N-terminales et
3 pour les extrêmités C-terminales avec les deux types d'acides aminés classiques Proline et Glycine.
Seuls deux I-sites sont clairement attachés aux
feuillets
avec 3 I-sites pour la partie centrale,
1 pour les N-terminales et
3 pour les extrêmités C-terminales avec les deux types d'acides aminés classiques Proline et Glycine.
Seuls deux I-sites sont clairement attachés aux
feuillets ![]() . Les 4 I-sites associés aux boucles représentent des
changements brusques et courts. Ce dernier fait est peut-ętre dû
ŕ l'élimination des boucles les plus longues de l'apprentissage.
. Les 4 I-sites associés aux boucles représentent des
changements brusques et courts. Ce dernier fait est peut-ętre dû
ŕ l'élimination des boucles les plus longues de l'apprentissage.
Le second point est l'absence de données concernant la structure des I-sites. Leur variabilité n'est pas clairement exprimée et ne concerne que le résultat de la prédiction.
La méthode se base sur les profils moyens obtenus précédement. Les scores obtenus sont pondérés en fonction de ceux obtenus dans la base d'apprentissage. Il convient de s'arréter particuličrement sur la méthode de calcul du taux de prédiction. Elle consiste ŕ calculer le I-site le plus probable pour un fragment de 8 résidus consécutifs. Ensuite il convient de comparer les angles dičdres du fragment avec ceux du I-site et, si la dma est inférieure ŕ 120o, les 8 sites sont considérés comme correctement prédits :
Une utilisation conjointe de PHD [165] avec les I-sites est possible, principalement pour trouver des séquences homologues. L'évaluation par PHD n'est pas une simple prédiction par type de structures secondaires, mais une redéfinition des angles des I-sites selon la concordance entre la prédiction de PHD et des I-sites.
55 nouvelles protéines sont testées et un taux de prédiction global de 48% avec les I-sites est trouvé, et un taux de 54% en combinant leur méthode avec la prédiction des structures secondaires par PHD. Le tableau 2.8 montrent les différents taux de prédiction selon le type de protéines avec les I-sites seuls, avec la méthode PHD et les deux combinées.
J'ai recalculé un pourcentage de prédiction modifié P' pour voir la sensibilité de leur métrique :
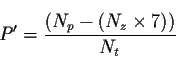
avec Np le nombre de sites correctement prédits, Nz, le nombre de zones continues de bonne prédiction et Nt le nombre total de résidus dans la protéine. Le principe est fort simple, si tous les sites correctement prédits sont continus, une seule zone existe donc. Alors, Nz=1, et seuls 7 sites sur les bords doivent être soustraits. La figure 2.22 montre le résulat pour une protéine de 100 résidus de longueur. La descente est moins accentuée avec une protéine de taille supérieure, mais l'exemple me parait frappant. Sur la figure, le dernier point hyp(1) représente l'hypothčse selon laquelle tous les I-sites correctement prédits sont indépendants et aucun chevauchement n'existe.
Le calibrage de ce type de fonction est complexe, car aucune idée du nombre de zones de I-sites correctement déterminées n'est accessible, en se réferant aux structures secondaires, il serait possible d'évaluer un nombre approximatif. Un calcul rapide montre un nombre de zones au minimum supérieur ŕ 7.
Les résultats récapitulés dans le tableau 2.8 montrent que les
hélices ![]() malgré leur importance dans les I-sites sont moyennement prédites, mais
que les feuillets
malgré leur importance dans les I-sites sont moyennement prédites, mais
que les feuillets ![]() ont un taux supérieur à celui généralement observé.
ont un taux supérieur à celui généralement observé.
Avant même la publication de l'article des I-sites [19], Baker et Bystroff les ont utilisés pour prédire des structures protéiques lors de CASP 2 [18]. Sur les 8 séquences, seules 2 ont été traitées par les I-sites combinés avec PHD. Les autres n'ont utilisé que I-sites. Cela ne change pas les taux qui sont de 39% pour les protéines avec les I-sites et 41% avec la méthode combinée.
Ils appliquent, ensuite, leur méthode ŕ la modélisation d'une zone dans un domaine SH3 [206]. Les scores de confiance sont particuličrement élevés dans cette zone. De plus, des données RMN sur la structure corroborrent leur application. Cet exemple pose néanmoins un autre problème: l'influence de l'alignement. Leurs I-sites fort bien prédits sont les parties les plus conservées, d'un point de vue séquence. Dans le même laboratoire, Simons et collaborateurs ont mis au point une méthode de modélisation ab initio sur un principe de statistique bayésienne avec recuit simulé [179]. Dans leur modčle bayésien le principe de relation séquence-structure vu par Han et Baker [75] est utilisé. Comme dans d'autres méthodes ab initio, seules les protéines de petites tailles ont un repliement acceptable. Ils essayent par la suite une série d'améliorations dont l'utilisation des I-sites, comme filtre au même titre que les structures secondaires, ce qui n'augmente pas l'efficacité de la méthode [180]. Pour CASP 3, l'utilisation de d'une nouvelle approche utilisant les I-sites et la méthode ab initio, travaillée cette fois-ci indépendament, a donné quelques trčs bons résultats [178].
Enfin, Bystroff et collaborateurs ont utilisé les I-sites pour prédire par une méthode de Chaînes de Markov Cachées les structures secondaires (nommé HMMSTR) avec un taux final de 74,3% [20]. Pour ceci, ils ont utilisé les 13 I-sites [19], plus trois nouveaux et ensuite ont effectué leur recherche en utilisant les angles dièdres pour catégoriser le type de structure prédite. Un détail intéressant est le passage du taux de prédiction des I-sites de 48 ŕ 59%, alors que le nombre de sites augmente, ce qui est surprenant. La figure 2.23 montre le calcul du taux de prédiction modifié P' dans ce nouveau cas. En réalité, avec quelques zones distinctes, le taux de prédiction est partiquement identique, ce qui peut expliquer ce taux "élevé" de prédiction.
(c) 2001- Alexandre de Brevern